This program counts the identical sequences and shows unique
sequences descending order.
This program also estimates Shannon-Weaver diversity index (H).
CDsearch performs RPS-BLAST search against NCBI CDD database.
Subsequences of the homologous region of the most conserved domain
for each of the input sequence will be extracted.
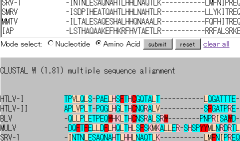
Alignment with clustalw or mafft.
You can now align nucleic sequences considering amino acid frame.
Select "tranalign" mode. This mode will translate the input sequence and align using mafft auto mode. Finally, the alignment will be refrected to the nucleotide sequence by tranaglign program.